Гемлібра (Hemlibra) – Еміцизумаб для профілактики гемофілії А
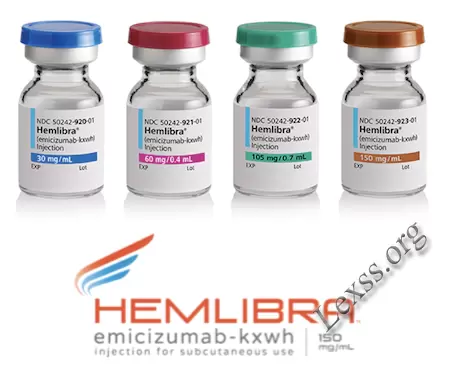
Гемлібра (Hemlibra)
Власник реєстраційного посвідчення:
F.Hoffmann-La Roche, Ltd (Швейцарія)
Вироблено та розфасовано:
CHUGAI PHARMA MANUFACTURING, Co.Ltd. (Японія) або SAMSUNG BIOLOGICS, Co.Ltd. (Республіка Корея)
Упаковка та контроль якості:
F.Hoffmann-La Roche, Ltd (Швейцарія) або ДОБРОЛЕК, ТОВ (Росія)
Контакти для звернень:
Ф. Хоффманн-Ля Рош Лтд. (Швейцарія)
Код ATX:
B02BX06
Активна речовина:
Еміцизумаб (emicizumab) Rec.INN зареєстрована ВООЗ
Торгове найменування:
Гемлібра (Hemlibra)
Міжнародне непатентоване найменування:
Еміцізумаб
Лікарська форма:
Розчин для підшкірного введення
Форма випуску, упакування та склад препарату Гемлібра:
Розчин для підшкірного введення у вигляді прозорої або опалесції, від безбарвного до жовтуватого кольору рідини.1 фл. (1 мл) еміцизумаб: 30 мг
Допоміжні речовини: L-гістидин – 3.1 мг, L-аспарагінова кислота – до рН 6.0, L-аргінін – 26.1 мг, полоксамер 188 – 0.5 мг, вода д/і – до 1 мл.
1 мл - флакони безбарвного скла (1) - картонні пачки×.
Розчин для підшкірного введення у вигляді прозорої або опалесції, від безбарвного до жовтуватого кольору рідини.1 фл. (0.4 мл) еміцизумаб: 60 мг
Допоміжні речовини: L-гістидин – 1.2 мг, L-аспарагінова кислота – до рН 6.0, L-аргінін – 10.5 мг, полоксамер 188 – 0.2 мг, вода д/і – до 0.4 мл.
0.4 мл - флакони безбарвного скла (1) - картонні пачки×.
Розчин для підшкірного введення у вигляді прозорої або опалесції, від безбарвного до жовтуватого кольору рідини.1 фл. (0.7 мл) еміцизумаб: 105 мг
Допоміжні речовини: L-гістидин – 2.2 мг, L-аспарагінова кислота – до рН 6.0, L-аргінін – 18.3 мг, полоксамер 188 – 0.4 мг, вода д/і – до 0.7 мл.
0.7 мл - флакони безбарвного скла (1) - картонні пачки×.
Розчин для підшкірного введення у вигляді прозорої або опалесції, від безбарвного до жовтуватого кольору рідини.1 фл. (1 мл) еміцизумаб: 150 мг
Допоміжні речовини: L-гістидин – 3.1 мг, L-аспарагінова кислота – до рН 6.0, L-аргінін – 26.1 мг, полоксамер 188 – 0.5 мг, вода д/і – до 1 мл.
1 мл - флакони безбарвного скла (1) - картонні пачки×.
× З метою контролю першого розтину на упаковку наноситься захисна голографічна наклейка.
На пачку наносяться захисні етикетки контролю першого відкриття (у разі упаковки на ТОВ "Добролік").
Опис:
Прозора або опалесцентна, від безбарвного до жовтуватого кольору рідина.
Клініко-фармакологічна група:
Моноклональні антитіла. Гемостатичний препарат
Фармако-терапевтична група:
Антитіла моноклональні
Фармакологічна дія:
Механізм дії:
Еміцизумаб є біспецифічними гуманізованими моноклональними антитілами на основі імуноглобуліну G4 (IgG4), що продукуються клітинами яєчників китайського хом'ячка за технологією рекомбінантної ДНК.
Еміцизумаб пов'язує активований фактор IX з фактором X для заповнення функції відсутнього активованого фактора VIII, який необхідний ефективного гемостазу.
Еміцизумаб не має структурної подібності або гомологічних послідовностей з фактором VIII (FVIII) і, відповідно, не індукує та не посилює утворення прямих інгібіторів FVIII.
Гемофілія А - це зчеплене з Х-хромосомою спадкове порушення зсідання крові внаслідок функціонального дефіциту FVIII, що призводить до крововиливів у суглоби, м'язи або внутрішні органи, спонтанним або при випадкових травмах, а також хірургічних втручань. Профілактика препаратом Гемлібра® вкорочує АЧТВ та збільшує показник активності FVIII, який визначається за хромогенним методом з використанням інших людських факторів згортання.
Дані фармакодинамічні маркери не відображають справжній гемостатичний ефект еміцизумабу in vivo (АЧТП надмірно вкорочено, показник активності FVIII може бути завищений), однак вони вказують на наявність у еміцизумабу прокоагулянтного ефекту.
Імуногенність:
При застосуванні препарату Гемлібра можливий розвиток імунної відповіді. Згідно з об'єднаними даними клінічних досліджень ІІІ фази у 668 пацієнтів проводили аналіз на наявність антитіл до еміцизумабу, при цьому у 34 (5.1%) пацієнтів результат був позитивним. У 18 (2,7%) пацієнтів антитіла до еміцизумабу були нейтралізуючими в умовах in vitro. У 14 з цих пацієнтів нейтралізуючі антитіла до еміцизумабу не мали клінічно значущого впливу на фармакокінетику або ефективність препарату Гемлібра®, при цьому у 4 (0.6%) пацієнтів відзначалося зниження концентрації еміцизумабу. У 1 (0.2%) пацієнта з нейтралізуючими антитілами до еміцизумабу та одночасним зниженням концентрації еміцизумабу спостерігалася втрата ефективності після 5 тижнів терапії препаратом Гемлібра®. Цей пацієнт припинив лікування препаратом Гемлібра®.
Загалом профіль безпеки препарату Гемлібра® у пацієнтів з антитілами до еміцизумабу (в т.ч. нейтралізуючими) відповідав такому у пацієнтів без антитіл до еміцизумабу (див. розділи "Побічна дія" та "Особливі вказівки"). Результати аналізу імуногенності, зокрема, кількість пацієнтів з позитивним результатом ІФА на антитіла до еміцизумабу (ELISA), та/або хромогенного аналізу активності FVIII на нейтралізуючі антитіла до еміцизумабу можуть залежати від різних факторів, таких як чутливість та специфічність аналізу, маніпуляції. , час забору зразків, супутні препарати та характер основного захворювання Виходячи з цих міркувань, порівняння частоти виявлення антитіл до еміцизумабу та частоти виявлення антитіл до інших біологічних препаратів може виявитися неінформативним.
Доклінічні дані з безпеки:
Доклінічні дослідження не виявили специфічних ризиків для людини.
Канцерогенність, генотоксичність, репродуктивна токсичність.
Чи не вивчалися.
Вплив на фертильність:
Еміцизумаб не викликав будь-яких токсикологічних змін у репродуктивних органах самців і самок яванських макак при введенні в дозі до 30 мг/кг/тиждень тривалістю до 26 тижнів, а також при внутрішньовенному введенні в дозах до 100 мг/кг/ тиждень протягом 4 тижнів.
Інше:
Рівні цитокінів, що вивільняються під дією еміцизумабу, були аналогічні таким для інших антитіл, що мають низький ризик індукції цитокінів, у дослідженні in vitro з використанням цілісної крові здорових дорослих добровольців.
Фармакокінетика:
Фармакокінетику еміцизумабу визначали за допомогою некомпартментного аналізу даних, одержаних у здорових добровольців, та популяційного фармакокінетичного аналізу даних, одержаних у пацієнтів з гемофілією А.
Всмоктування:
Період напів всмоктування після підшкірного введення препарату пацієнтам з гемофілією А становив 1.6 днів.
Середні (± стандартне відхилення (СО)) мінімальні концентрації еміцизумабу в плазмі досягали значень 52.6±13.6 мкг/мл на 5-му тижні після багаторазових підшкірних ін'єкцій препарату в дозі 3 мг/кг 1 раз на тиждень пацієнтам з гемофілією А. Середні мінімальні концентрації еміцизумабу в плазмі в рівноважному стані становили 51.2 мкг/мл, 46.9 мкг/мл і 38.5 мкг/мл при застосуванні препарату в підтримуючій дозі 1.5 мг/кг 1 раз на тиждень, 3 мг/кг 1 раз на 2 мг/кг 1 раз на 4 тижні, відповідно (див. малюнок 1 та таблицю 1).
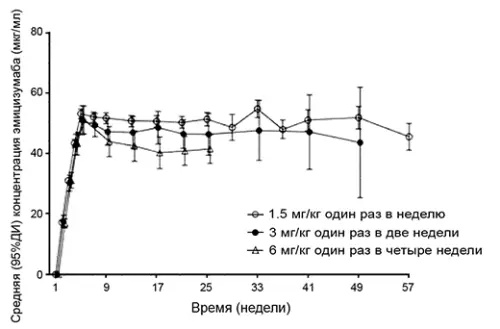
Середні (±СО) мінімальна концентрація (Ctrough), Cmax та відношення Cmax/Ctrough у рівноважному стані при застосуванні рекомендованих підтримуючих доз 1.5 мг/кг 1 раз на тиждень, 3 мг/кг 1 раз на 2 тижні та 6 мг/кг 1 раз у 4 тижні представлені у таблиці 1.
| Параметр | Підтримуюча доза | ||
| 1.5 мг/кг 1 раз на тиждень | 3 мг/кг 1 раз на 2 тижні | 6 мг/кг 1 раз на 4 тижні | |
| Cmax,ss (мкг/мл) | 55.1±5.9 | 58.3±16.4 | 67.0±17.7 |
| Cavg,ss (мкг/мл) | 53.7±15.6 | 53.7±15.6 | 53.7±15.6 |
| Ctrough,ss (мкг/мл) | 51.2±15.2 | 46.9±14.8 | 38.5±14.2 |
| Співвідношення Cmax/Ctrough | 1.08±0.03 | 1.26±0.12 | 1.85±0.47 |
Cavg,ss = середня концентрація у рівноважному стані.
Cmax,ss = максимальна концентрація у плазмі у рівноважному стані.
Ctrough, ss = мінімальна концентрація у рівноважному стані.
Фармакокінетичні параметри одержані з популяційної фармакокінетичної моделі.
У пацієнтів ≥12 років (дорослі/підлітки) та <12 років (діти) після застосування препарату в дозі 3 мг/кг 1 раз на тиждень протягом 4 тижнів, а потім у підтримуючій дозі 1.5 мг/кг 1 раз на тиждень спостерігалися подібні профілі фармакокінетики (рис. 2).
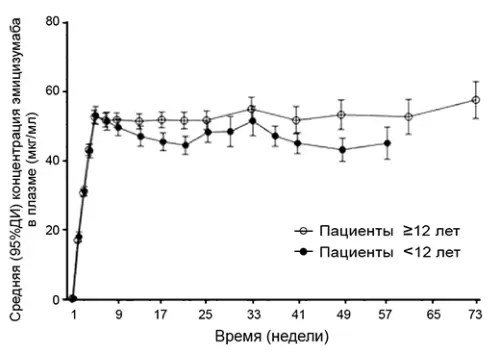
У здорових добровольців абсолютна біодоступність після введення препарату в дозі 1 мг/кг варіювала від 80.4% до 93.1% залежно від місця введення. Профілі фармакокінетики після підшкірного введення еміцизумабу в ділянку живота, верхню частину зовнішньої поверхні плеча та стегно були подібними. Препарат можна вводити по черзі у зазначені області.
Розподіл:
Після одноразового внутрішньовенного введення еміцизумабу в дозі 0.25 мг/кг здоровим добровольцям Vd у рівноважному стані становив 106 мл/кг (тобто 7.4 л для дорослої людини з масою тіла 70 кг). Препарат не призначений для внутрішньовенного введення.
У пацієнтів з гемофілією А після багаторазових підшкірних ін'єкцій здається Vd, розрахований згідно з даними популяційного фармакокінетичного аналізу, склав 10.4 л.
Лінійність дози. Еміцизумаб демонстрував пропорційну дозі фармакокінетику у пацієнтів з гемофілією А при підшкірному введенні в діапазоні доз 0.3-6 мг/кг 1 раз на тиждень.
Метаболізм:
Метаболізм еміцизумабу не вивчався. Антитіла IgG переважно каталізуються шляхом лізосомального протеолізу, потім продукти розпаду антитіл (амінокислоти) виводяться або використовуються організмом.
Виведення:
Після внутрішньовенного введення еміцизумабу в дозі 0.25 мг/кг здоровим добровольцям загальний кліренс становив 3.26 мл/кг/добу (тобто 0.228 л/добу у дорослого з масою тіла 70 кг); середній Т1/2 – 26.7 днів.
Після одноразового підшкірного введення здоровим добровольцям Т1/2 становив приблизно 4-5 тижнів.
Після багаторазових підшкірних ін'єкцій препарату пацієнтам з гемофілією А кліренс, що здається, склав 0.271 л/добу, що здається Т1/2 - 26.9 днів.
Фармакокінетика у пацієнтів спеціальних груп:
Пацієнти із порушенням функції нирок:
Безпека та ефективність препарату Гемлібра® у пацієнтів з порушенням функції нирок не вивчалися окремо.
Доступні дані щодо застосування препарату Гемлібра у пацієнтів з порушенням функції нирок легкого та середнього ступеня тяжкості обмежені.
Дані щодо застосування препарату Гемлібра у пацієнтів з порушенням функції нирок тяжкого ступеня відсутні.
Препарат Гемлібра® є моноклональним антитілом і виводиться з організму шляхом катаболізму, а не нирками.
Спеціальних досліджень щодо вивчення впливу порушення функції нирок на фармакокінетику еміцизумабу не проводилося. Більшість пацієнтів з гемофілією А, які увійшли до популяційного аналізу фармакокінетики, мали нормальну функцію нирок (КК ≥90 мл/хв) або порушення функції нирок легкого ступеня тяжкості (КК 60-89 мл/хв). Тільки два пацієнти мали порушення функції нирок середнього ступеня тяжкості (КК 30-59 мл/хв). Жоден пацієнт не мав порушення функції нирок тяжкого ступеня. Порушення функції нирок легкого або середнього ступеня тяжкості не впливало на фармакокінетику еміцизумабу.
Таким чином, вважається, що зміна дози у пацієнтів з порушенням функції нирок не потрібна.
Пацієнти з порушенням функції печінки:
Безпека та ефективність препарату Гемлібра® у пацієнтів з порушенням функції печінки не вивчалися окремо.
Пацієнти з порушенням функції печінки легкого та середнього ступеня тяжкості брали участь у клінічних дослідженнях.
Дані щодо застосування препарату Гемлібра у пацієнтів з порушенням функції печінки тяжкого ступеня відсутні.
Препарат Гемлібра® є моноклональним антитілом і виводиться з організму шляхом катаболізму, а не шляхом печінкового метаболізму.
Спеціальних досліджень щодо вивчення впливу порушення функції печінки на фармакокінетику еміцизумабу не проводилося.
Більшість пацієнтів з гемофілією А, що увійшли до популяційного аналізу фармакокінетики, мали нормальну функцію печінки (білірубін та ACT ≤ ВГН) або порушення функції печінки легкого ступеня тяжкості (білірубін ≤ВГН та ACT >ВГН або білірубін <1.0-15 ×). ).
Тільки у шести пацієнтів було порушення функції печінки середнього ступеня тяжкості (1.5 ВГН <білірубін ≤ 3 ВГН і будь-яка активність ACT).
Порушення функції печінки легкого або середнього ступеня тяжкості не впливало на фармакокінетику еміцизумабу.
Порушення функції печінки визначали згідно з критеріями Національного Інституту Онкології для дисфункції печінки.
Таким чином, вважається, що зміна дози у пацієнтів з порушенням функції печінки не потрібна.
Пацієнти дитячого віку:
Вплив віку на фармакокінетику еміцизумабу оцінювали у популяційному фармакокінетичному аналізі у немовлят (від ≥1 місяця до <2 років), дітей (від ≥2 до <12 років) та підлітків (від 12 до <18 років) з гемофілією А. Вік не чинив впливу на фармакокінетику еміцизумабу.
Пацієнти похилого віку:
Вплив віку на фармакокінетику еміцизумабу оцінювали в фармакокінетичному популяційному аналізі, що включав пацієнтів ≥65-<77 років. Відносна біодоступність препарату знижувалась зі збільшенням віку, проте клінічно значимих відмінностей у фармакокінетиці еміцизумабу у пацієнтів <65 років та пацієнтів ≥65 років не було. Безпека та ефективність застосування препарату Гемлібра® у пацієнтів похилого віку окремо не вивчалися. Клінічні дослідження препарату Гемлібра включали пацієнтів віком 65 років.
Раса:
Популяційний фармакокінетичний аналіз у пацієнтів із гемофілією А показав, що раса не впливає на фармакокінетику еміцизумабу.
Показання для застосування Гемлібра:
Як рутинна профілактика для запобігання або зниження частоти кровотеч у пацієнтів з:
- Гемофілією А (спадковий дефіцит фактора VIII) з інгібіторами фактора VIII;
- Тяжкою формою гемофілії А (спадковий дефіцит фактора VIII, FVIII <1%) без інгібіторів фактора VIII.
Режим дозування:
Терапію слід розпочинати під наглядом лікаря, який має досвід у лікуванні гемофілії та/або порушень згортання крові.
Лікування препаратами із шунтуючим механізмом дії (bypassing agents) слід припинити за день до початку терапії препаратом Гемлібра®. Профілактику фактором VIII можна продовжувати протягом перших 7 днів терапії препаратом Гемлібра.
Рекомендований режим дозування:
Рекомендована доза становить 3 мг/кг у вигляді підшкірної ін'єкції 1 раз на тиждень протягом перших 4 тижнів, потім препарат вводять у підтримуючій дозі:
- 1.5 мг/кг 1 раз на тиждень або
- 3 мг/кг 1 раз на 2 тижні або
- 6 мг/кг 1 раз на 4 тижні.
Спосіб застосування:
Препарат Гемлібра призначений лише для підшкірного введення.
Препарат слід запроваджувати з дотриманням належних правил асептики.
Вибір місця для ін'єкції слід обмежити рекомендованими ділянками:
- Область живота,
- Верхня частина зовнішньої поверхні плеча
- Стегно.
Дані про ін'єкції інші ділянки тіла відсутні.
П/к ін'єкції препарату Гемлібра у верхню частину зовнішньої поверхні плеча повинні виконуватися особою, яка здійснює догляд за пацієнтом, або медичним працівником.
Чергування місць ін'єкцій може допомогти запобігти або зменшити реакції у місці введення.
Не слід вводити препарат Гемлібра у:
- Родині плями;
- Тканини рубців;
- Гематоми;
- Місця з ущільненням або пошкодженням;
- Ділянки з чутливою шкірою;
- Почервонінням.
Препарат Гемлібра та інші препарати, також призначені для підшкірного введення, переважно вводити в різні анатомічні області.
Введення препарату пацієнтом та/або особою, яка здійснює догляд за пацієнтом
Препарат Гемлібра призначений для застосування під керівництвом медичного працівника. Після належного навчання техніці підшкірних ін'єкцій пацієнт може вводити препарат Гемлібра самостійно. На розсуд лікаря препарат Гемлібра може вводитися особою, яка здійснює догляд за пацієнтом.
Лікар та особа, яка здійснює догляд за пацієнтом, повинні оцінити можливість самостійного введення препарату Гемлібра дитиною. Однак самостійне введення препарату дітьми до 7 років не рекомендовано.
Тривалість лікування:
Препарат Гемлібра призначений для довгострокової профілактики.
Корекція дози:
Корекція дози препарату Гемлібра не рекомендується.
Затримка прийому чи пропуск дози:
Якщо пацієнт пропустив планову підшкірну ін'єкцію препарату Гемлібра, його слід проінструктувати ввести пропущену дозу якнайшвидше, до дня введення чергової планової дози. Потім пацієнту слід запровадити наступну дозу у звичайний запланований день введення. Пацієнт не повинен отримувати дві дози на один день для заповнення пропущеної дози.
Дозування в особливих випадках:
- Корекція дози у пацієнтів дитячого віку не потрібна.
- Корекція дози у пацієнтів похилого віку (≥65 років) не потрібна.
- Корекція дози у пацієнтів з порушенням функції нирок та пацієнтів з порушенням функції печінки не потрібна.
Поводження з препаратом:
Препарат Гемлібра® являє собою стерильний розчин, що не містить консервантів, готовий до використання і не вимагає розведення розчину для підшкірного введення, від безбарвного до блідо-жовтого кольору.
Перед введенням слід візуально перевірити розчин на предмет механічних включень та зміни забарвлення.
За наявності видимих механічних включень або зміни забарвлення препарат не можна використовувати і необхідно утилізувати.
Флакони препарату Гемлібра у лікарській формі розчин для підшкірного введення призначені тільки для одноразового застосування.
Препарат Гемлібра слід зберігати в холодильнику (2-8°С). Після виймання з холодильника нерозкриті флакони можна зберігати при кімнатній температурі (нижче 30°С) трохи більше 7 днів.
Після зберігання при кімнатній температурі нерозкриті флакони можуть бути знову поміщені в холодильник. Загальний сумарний час зберігання препарату за кімнатної температури не повинен перевищувати 7 днів.
Посібник з використання препарату:
Для вилучення препарату Гемлібра з флакону та його підшкірного введення необхідні шприц, голка для перенесення, ін'єкційна голка, які відповідають наступним критеріям.
Шприц 1 мл:
Прозорий поліпропіленовий або полікарбонатний, одноразовий, ін'єкційний, безлатексний, апірогенний, стерильний шприц з канюлею Луер-Лок (у разі, якщо шприц з канюлею Луєр-Лок недоступний, може бути використаний шприц з канюлею Луер-Сліп) та градуюванням.
Шприц 2-3 мл:
Прозорий поліпропіленовий або полікарбонатний, одноразовий, ін'єкційний, безлатексний, апірогенний, стерильний шприц з канюлею Луер-Лок (у випадку, якщо шприц з канюлею Луєр-Лок недоступний, може бути використаний шприц з канюлею Луер-Сліп) та градуюванням.
Голка для перенесення:
Стерильна, безлатексна, апірогенна, одноразова голка з нержавіючої сталі зі з'єднанням Луер-Лок (у разі, якщо голка зі з'єднанням Луер-Лок недоступна, може бути використана голка зі з'єднанням Луер-Сліп) калібру 18G, з довжиною 26 мм (1'' )-40 мм (1.5'').
Ін'єкційна голка:
Стерильна, безлатексна, апірогенна, одноразова голка з нержавіючої сталі зі з'єднанням Луер-Лок (у разі, якщо голка зі з'єднанням Луер-Лок недоступна, може бути використана голка зі з'єднанням Луер-Сліп) калібру 26G (прийнятний діапазон: 25-27G), з довжиною 9 мм (3/8'') (переважно) або 13 мм (1/2'') (максимально), із запобіжником (переважно) або без нього.
Для введення препарату об'ємом ≤1 мл слід використовувати Шприц 1 мл, для введення препарату об'ємом >1 мл та ≤2 мл слід використовувати Шприц 2-3 мл. Не слід вводити об'єм препарату >2 мл за одну ін'єкцію.
Якщо для введення призначеної дози потрібне вилучення препарату з декількох флаконів в один шприц, див. нижче інформацію (підрозділ "Об'єднання флаконів"). Не можна поєднувати флакони, що містять препарат у різній концентрації, в один шприц. Після перенесення з флакона до шприца лікарський препарат слід використовувати негайно, т.к. він не містить антимікробних консервантів.
Підготовка до використання препарату:
- Перед застосуванням необхідно залишити флакон нагрітися при кімнатній температурі протягом приблизно 15 хв на чистій плоскій поверхні подалі від прямих сонячних променів.
- Не слід намагатися зігрівати флакон будь-яким іншим способом.
- Вимити руки водою з милом.
Вибір та підготовка місця ін'єкції:

- Ретельно обробити намічене місце ін'єкції спиртовою серветкою.
- Зачекати приблизно 10 сек, поки оброблена ділянка підсохне.
- Не торкайтеся цієї області до ін'єкції.
- Забороняється обмахувати або обдувати очищену ділянку.
Ін'єкції рекомендується проводити:
- У передню та середню поверхню стегна;
- У нижню частину живота, за винятком області діаметром 5 см безпосередньо навколо пупка;
- У верхню частину зовнішньої поверхні плеча (тільки якщо ін'єкція виробляється особою, яка доглядає пацієнта).
Необхідно щоразу змінювати місце ін'єкції (при проведенні ін'єкції рекомендується відступати не менше ніж на 2.5 см від попередньої ін'єкції).
Слід уникати ділянок, які можуть роздратуватися ременем або поясом одягу. Не слід вводити препарат у рідні плями, тканини рубців, гематоми, місця з ущільненням, пошкодженням, ділянки з чутливою шкірою, почервонінням.
Важлива інформація про поводження зі шприцом:
- Після видалення ковпачка не слід торкатися голок і класти їх на будь-яку поверхню.
- Після заповнення шприца розчином препарату він має бути використаний негайно.
- Підшкірна ін'єкція повинна бути завершена не пізніше ніж через 5 хв після видалення ковпачка з ін'єкційної голки. Не слід вводити препарат, якщо торкнулися голкою будь-якої поверхні.
- Інструкції з утилізації використаних шприців див. у розділі "Рекомендації з утилізації".
Важлива інформація після ін'єкції:
- Не слід розтирати місце введення після ін'єкції.
- Якщо після завершення ін'єкції з'явилися краплі крові, слід натиснути стерильним ватним або марлевим тампоном на місце ін'єкції, як мінімум, протягом 10 секунд, поки кровотеча не припиниться.
- Якщо виникла гематома (невелика підшкірна кровотеча), слід також прикласти пакет із льодом та злегка натиснути на цю ділянку.
Якщо кровотеча не зупиняється, пацієнту слід звернутися до медичного працівника.
Підготовка до введення препарату:
Крок 1. Зняття ковпачка та очищення верхньої частини флакона.

- Зняти ковпачок із флакона.

- Очистити верхню частину пробки флакона спиртовою серветкою.
- Помістити ковпачок від флакона у захищений від проколів контейнер.
Крок 2. Приєднання голки для перенесення до шприца*

- Натиснути та повертати голку для перенесення за годинниковою стрілкою доти, доки вона не буде повністю приєднана до шприца.
* Тут і далі на малюнках представлені шприц та голки зі з'єднанням Луєр-Лок. Однак, якщо шприц та/або голки зі з'єднанням Луєр-Лок недоступні, можуть бути використані шприц та/або голки зі з'єднанням Луєр-Сліп. У разі використання шприца та/або голок зі з'єднанням Луер-Сліп також див.

Повільно відтягнути поршень і набрати ту кількість повітря, яка дорівнює обсягу призначеної дози.
Крок 3. Зняття ковпачка з голки для перенесення

- Тримати шприц за циліндр, при цьому голка для перенесення має бути спрямована вгору.
- Обережно потягнути за ковпачок голки для перенесення. Рух має бути спрямований від себе. Чи не викидати ковпачок, слід покласти його на плоску поверхню. Необхідно надіти ковпачок на голку для перенесення після вилучення препарату із флакона.
- Не торкатися кінця голки і не поміщати її на будь-яку поверхню після зняття ковпачка.
Крок 4. Введення повітря у флакон

- Поставити флакон на плоску робочу поверхню і ввести голку для перенесення, приєднану до шприца безпосередньо в центр пробки флакона.

Перевернути флакон разом з голкою, що знаходиться в ньому.

- Направити голку вгору і натиснути на поршень, щоб випустити повітря зі шприца поверх розчину.
- Утримувати палець натиснутим на поршні шприца.
- Не вводити повітря безпосередньо в розчин, оскільки це може призвести до утворення бульбашок повітря.
Крок 5. Перенесення препарату в шприц

- Опустити кінець голки так, щоб він був у розчині.
- Повільно тягнути поршень для заповнення шприца об'ємом розчину більше за призначену дозу.
- Слід бути обережними, щоб не витягнути поршень зі шприца.
Важливо! Якщо обсяг призначеної дози більший за об'єм препарату у флаконі слід витягти весь розчин з флакона (див. підрозділ "Об'єднання флаконів").
Крок 6. Видалення бульбашок повітря
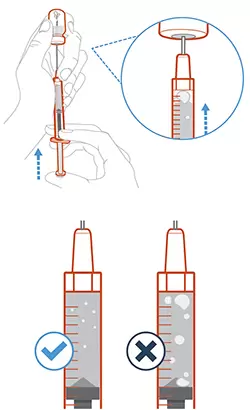
- Утримуючи голку у флаконі, перевірити шприц щодо великих бульбашок повітря. Дуже великі бульбашки повітря можуть зменшити дозу, яку необхідно отримати.
- Видалити великі бульбашки повітря таким чином: слід акуратно постукувати пальцями по циліндру шприца, поки бульбашки повітря не перемістяться у верхню частину шприца. Перемістити кінець голки поверх розчину і повільно натиснути на поршень для видалення бульбашок повітря зі шприца.
- Якщо об'єм препарату в шприці дорівнює або менший за призначену дозу, перемістити кінець голки, щоб він знаходився в розчині, і повільно відтягувати поршень доти, поки в шприц не буде набраний об'єм препарату більше призначеної дози.
- Необхідно бути обережними, щоб не витягнути поршень зі шприца.
- Слід повторювати вищезгадані кроки до тих пір, поки не будуть видалені всі великі бульбашки повітря.
Примітка: Необхідно переконатися, що набрано достатню кількість препарату в шприц для отримання призначеної дози перед тим, як перейти до наступного кроку. Якщо не вдається витягнути розчин з флакона повністю, слід перевернути флакон вертикально, щоб набрати кількість, що залишилася.
Не можна використовувати голку для перенесення, щоб зробити ін'єкцію, т.к. це може завдати шкоди, а саме, призвести до виникнення кровотечі та болю.
Введення препарату:
Крок 7. Закриття ковпачка на голці для перенесення

- Витягти голку для перенесення, приєднану до шприца, із флакона.
- Однією рукою засунути голку для перенесення в ковпачок і підняти зачерпним рухом таким чином, щоб ковпачок закривав голку для перенесення.
- Після цього натиснути однією рукою на ковпачок у напрямку до шприца, щоб повністю приєднати його, щоб запобігти випадковому уколу голкою.
Крок 8. Обробка місця ін'єкції
- Вибрати місце ін'єкції та обробити його спиртовою серветкою.
Крок 9. Видалення голки для перенесення

- Для вилучення голки для перенесення зі шприца повернути її проти годинникової стрілки та обережно потягнути.
- Помістити використану голку для перенесення в захищений від проколів контейнер.
Крок 10. Приєднання ін'єкційної голки** до шприцу

- Натиснути та повертати ін'єкційну голку за годинниковою стрілкою доти, доки вона не буде повністю приєднана до шприца.
** Тут і далі на малюнках представлена ін'єкційна голка із запобіжником. Однак також допускається застосування ін'єкційної голки без запобіжника. У разі використання голки без запобіжника також див. відповідну інструкцію із застосування, розроблену виробником, щодо особливостей поводження з такою голкою.
Крок 11. Усунення запобіжника з ін'єкційної голки

- Зрушити запобіжник з ін'єкційної голки до циліндра шприца.
Крок 12. Зняття ковпачка з ін'єкційної голки

- Обережно потягнути ковпачок ін'єкційної голки у напрямку від шприца.
- Помістити ковпачок у захищений від проколів контейнер.
- Не торкатися кінця голки і не допускати дотику будь-якої поверхні з кінцем голки.
- Ін'єкція повинна бути завершена не пізніше ніж через 5 хв після видалення ковпачка з ін'єкційної голки.
Крок 13. Регулювання положення поршня до призначеної дози

- Повільно натискати на поршень для досягнення розподілу на шприці, що відповідає призначеній дозі.
- Переконайтеся, що верхня кромка поршня знаходиться врівні з розподілом на шприці, який відповідає призначеній дозі.
Крок 14. Підшкірна ін'єкція

- Стиснути місце ін'єкції та, не натискаючи на поршень, швидким твердим рухом повністю ввести голку у шкірну складку під кутом 45-90°.
- Утримуючи положення шприца, відпустіть місце ін'єкції.
Крок 15. Введення препарату

- Плавно натискаючи на поршень, повільно запровадити весь лікарський препарат.
- Витягти голку, приєднану до шприца, з місця ін'єкції під тим самим кутом, під яким вводилося.
Крок 16. Закриття ін'єкційної голки запобіжником

- Пересуньте запобіжник вперед на 90° у напрямку від циліндра шприца.
- Утримуючи шприц однією рукою, притиснути запобіжник до плоскої поверхні швидким рухом доти, доки не почується клацання.

- Якщо клацання не було, слід перевірити, чи закриває запобіжник голку повністю.
- Слід завжди тримати пальці за запобіжником і далеко від самої ін'єкційної голки.
- Не слід від'єднувати ін'єкційну голку.
Крок 17. Утилізація шприца та голки

- Помістити всі використані голки та шприци у захищений від проколів контейнер відразу після використання. Докладнішу інформацію див. у розділі "Рекомендації з утилізації".
- Не слід намагатися витягти використану ін'єкційну голку із використаного шприца.
- Не надягати повторно ковпачок на ін'єкційну голку.
Важливо: завжди зберігайте захищений від проколів контейнер у недоступному для дітей місці.
Об'єднання флаконів:
Якщо потрібно використовувати більше 1 флакона для отримання призначеної дози, див. нижченаведені кроки після вилучення препарату з першого флакона.
Крок А. Закриття ковпачка на голці для перенесення
- Витягти голку для перенесення, приєднану до шприца, із флакона.
- Однією рукою засунути голку для перенесення в ковпачок і підняти зачерпним рухом таким чином, щоб ковпачок закривав голку для перенесення.
- Після цього натиснути однією рукою на ковпачок у напрямку до шприца, щоб повністю приєднати його, щоб запобігти випадковому уколу голкою.
Крок Б. Видалення голки для перенесення
- Для вилучення голки для перенесення зі шприца повернути її проти годинникової стрілки та обережно потягнути.
- Помістити використану голку для перенесення в захищений від проколів контейнер.
Крок В. Приєднання нової голки для перенесення до шприца
Примітка: Необхідно використовувати нову голку для перенесення кожного разу, коли виймаєте розчин з нового флакона.
- Натиснути та повертати нову голку для перенесення за годинниковою стрілкою доти, доки вона не буде повністю приєднана до шприца.
- Повільно відтягнути поршень і набрати повітря у шприц.
Крок Г. Зняття ковпачка з голки для перенесення
- Слід тримати шприц за циліндр, при цьому голка для перенесення має бути спрямована вгору.
- Обережно потягнути за ковпачок голки для перенесення. Рух має бути спрямований від себе. Не викидати ковпачок; слід покласти його на пласку поверхню. Необхідно надіти ковпачок на голку для перенесення після вилучення препарату із флакона.
- Не торкатися до кінця голки.
Крок Ґ. Введення повітря у флакон
- Поставити флакон на плоску робочу поверхню та ввести голку для перенесення, приєднану до шприца, безпосередньо до центру пробки флакона.
- Перевернути флакон разом із голкою, що знаходиться в ньому.
- Направити голку вгору і натиснути на поршень, щоб випустити повітря зі шприца поверх розчину.
- Утримувати палець натиснутим на поршні шприца.
- Не вводити повітря безпосередньо в розчин, оскільки це може призвести до утворення бульбашок повітря.
Крок Д. Перенесення препарату в шприц
- Опустити кінець голки так, щоб він був у розчині.
- Повільно тягнути поршень для того, щоб заповнити шприц об'ємом розчину більше за об'єм призначеної дози.
- Слід бути обережними, щоб не витягнути поршень зі шприца.
Примітка: Перед тим, як перейти до наступного кроку, слід переконатися, що набрано достатню кількість препарату в шприц для отримання призначеної дози. Якщо не вдається витягнути розчин з флакона повністю, слід перевернути флакон вертикально, щоб набрати кількість, що залишилася.
Не можна використовувати голку для перенесення, щоб зробити ін'єкцію, т.к. це може завдати шкоди, а саме, призвести до виникнення кровотечі та болю.
Слід повторювати кроки А-Д з кожним додатковим флаконом доти, доки в шприц не буде набрано об'єм препарату більше за обсяг призначеної дози. Після цього, не виймаючи голку для перенесення з флакона, слід повернутися до кроку 6 і завершити кроки, що залишилися.
Рекомендації щодо утилізації:
Усі використані шприци, флакони, голки, ковпачки від флаконів та ін'єкційних голок повинні бути поміщені у захищений від проколів контейнер одразу після використання. Голки та шприци не можна використовувати повторно.
Не слід утилізувати відкриті голки та шприци з побутовими відходами.
Якщо захищений від проколів контейнер недоступний, можна використовувати контейнер для збирання твердих побутових відходів, який:
- Зроблений із міцного пластику;
- Може бути закритий щільною захищеною від проколів кришкою, щоб гострі предмети були ізольовані;
- Вертикальний та стійкий у ході використання;
- Герметичний;
- Належним чином промаркований для попередження про вміст небезпечних відходів.
Побічна дія:
Профіль безпеки оцінено виходячи з об'єднаних даних клінічних досліджень профілактики препаратом Гемлібра® у дорослих (≥18 років), підлітків (від ≥12 років до <18 років), дітей (від ≥2 років до <12 років) та немовлят (від ≥1 місяця до <2 років) чоловічої статі з гемофілією А. Медіана тривалість застосування препарату становила 34.1 тижня (0.1-94.3 тижні).
У 0.8% пацієнтів лікування препаратом Гемлібра було припинено внаслідок небажаних реакцій, а саме тромботичної мікроангіопатії, некрозу шкіри з одночасним тромбофлебітом поверхневих вен, а також головного болю. Небажані реакції згруповані відповідно до класів систем органів медичного словника для нормативно-правової діяльності MedDRA. Для опису частоти небажаних реакцій використовується така класифікація: дуже часто (≥10%), часто (≥1% та <10%), нечасто (≥0.1% та <1%).
Інфекційні та паразитарні захворювання: нечасто – тромбоз кавернозного синуса.
З боку крові та лімфатичної системи: нечасто – тромботична мікроангіопатія.
З боку нервової системи: дуже часто – головний біль.
З боку серцево-судинної системи: нечасто – тромбофлебіт поверхневих вен.
З боку шлунково-кишкового тракту: часто – діарея.
З боку шкіри та підшкірних тканин: нечасто – некроз шкіри.
З боку кістково-м'язової системи: дуже часто – артралгія; часто – міалгія.
Загальні розлади та порушення у місці введення: дуже часто - реакції у місці введення; часто – пірексія.
Опис окремих небажаних реакцій:
Найбільш серйозними небажаними реакціями, що спостерігалися у клінічних дослідженнях препарату Гемлібра, були тромботична мікроангіопатія (ТМА) та тромботичні явища, в т.ч. тромбоз кавернозного синуса та тромбофлебіт поверхневих вен з одночасним некрозом шкіри.
Тромботична мікроангіопатія:
ТМА спостерігалася у <1% пацієнтів та у 9.7% пацієнтів, які отримали як мінімум одну дозу аКПК у клінічних дослідженнях. Зазначалося, що кожен з цих пацієнтів перед розвитком явищ ТМА (які проявляються у вигляді тромбоцитопенії, мікроангіопатичної гемолітичної анемії та гострого пошкодження нирок при відсутності тяжкого дефіциту активності ADAMTS13 (металопротеїнази, що розщеплює фактор Віллебранда)) отримував середню 100 Од/кг/24 год протягом ≥24 годин одночасно з профілактикою препарату Гемлібра®. Один пацієнт відновив прийом препарату Гемлібра після дозволу ТМА без рецидиву.
Тромботичні явища:
Серйозні тромботичні явища спостерігалися у <1% пацієнтів та у 6.5% пацієнтів, які отримали як мінімум одну дозу аКПК у клінічних дослідженнях. Повідомлялося, що кожен із цих пацієнтів перед розвитком тромботичних явищ отримував середню кумулятивну дозу аКПК >100 Од/кг/24 години протягом ≥24 годин одночасно з профілактикою препарату Гемлібра®. Один пацієнт відновив прийом препарату Гемлібра після дозволу тромботичного явища без рецидиву.
Опис терапії аКПК (об'єднані дані клінічних досліджень):
Є дані про 82 випадки терапії аКПК*, з яких у 8 випадках (10%) пацієнти отримували середню кумулятивну дозу аКПК >100 Од/кг/24 год протягом ≥24 год; у двох із 8 випадків відзначали тромботичні явища, у трьох із 8 випадків - ТМА (див. таблицю 2). В інших випадках тромботичних явищ та ТМА не спостерігалося. З усіх випадків лікування аКПК 68% складалося з одноразової інфузії ≤100 Од/кг.
Таблиця 2. Опис терапії аКПК* (об'єднані дані клінічних досліджень)
| Тривалість застосування аКПК | Середня кумулятивна доза аКПК протягом 24 годин (Од/кг/24 год) | ||
| <50 | 50-100 | >100 | |
| <24 ч | 9 | 47 | 13 |
| 24-48 ч | 0 | 3 | 1а |
| >48 ч | 1 | 1 | 7а,б |
* Випадок терапії аКПК - усі дози аКПК, отримані пацієнтом з будь-якої причини, до моменту 36 годин перерви у лікуванні. Включає всі випадки терапії аКПК, за винятком введення аКПК у перші 7 днів застосування препарату Гемлібра® та після 30 днів з моменту його припинення.
- а Тромботичне явище.
- б Тромботична мікроангіопатія.
Реакції у місці введення:
У клінічних дослідженнях дуже часто (21%) спостерігалися реакції у місці введення, які були несерйозними, легкого та середнього ступеня тяжкості, та у 95% випадків вирішилися без лікування. Симптомами, про які повідомлялося часто, були еритема у місці введення (11%), біль у місці введення (4%) та свербіж у місці введення (3%).
Імуногенність:
Згідно з даними об'єднаних клінічних досліджень ІІІ фази препарату Гемлібра® нейтралізуючі антитіла до еміцизумабу з одночасним зниженням концентрації еміцизумабу виникали нечасто (див. розділ "Фармакологічна дія"). У одного пацієнта з нейтралізуючими антитілами до еміцизумабу та одночасним зниженням концентрації еміцизумабу відзначалася втрата ефективності (маніфестація у вигляді проривної кровотечі) після 5 тижнів терапії. Цей пацієнт припинив застосування препарату Гемлібра® (див. розділи "Фармакологічна дія" та "Особливі вказівки"). Загалом профіль безпеки препарату Гемлібра у пацієнтів з антитілами до еміцизумабу (в т.ч. нейтралізуючими) відповідав такому у пацієнтів без антитіл до еміцизумабу.
Протипоказання до застосування:
- Підвищена чутливість до еміцизумабу або до будь-якої допоміжної речовини в анамнезі;
- Вагітність (ефективність та безпека застосування не вивчалися);
- Період грудного вигодовування (ефективність та безпека застосування не вивчалися).
З обережністю: порушення функції нирок та печінки тяжкого ступеня.
Застосування при вагітності та годуванні груддю:
Контрацепція:
Жінкам із дітородним потенціалом слід використовувати ефективні способи контрацепції під час терапії препаратом Гемлібра® та протягом не менше 6 місяців після останнього прийому препарату.
Вагітність:
Клінічні дослідження вагітних жінок не проводилися. Вплив на репродуктивну функцію тварин не вивчалося. Невідомо, чи може препарат Гемлібра при застосуванні вагітними жінками чинити шкідливу дію на плід або впливати на репродуктивну здатність. Застосування препарату Гемлібра при вагітності протипоказане.
Період грудного вигодовування:
Невідомо, чи проникає еміцизумаб у грудне молоко. Дослідження щодо вивчення впливу еміцизумабу на утворення молока або його присутності в грудному молоці не проводилися. Людський IgG проникає у грудне молоко. Застосування препарату Гемлібра у період грудного вигодовування протипоказане.
Застосування при порушеннях функції печінки:
З обережністю слід призначати препарат при порушеннях функції печінки тяжкого ступеня.
Застосування при порушеннях функції нирок:
З обережністю слід призначати препарат при порушеннях функції нирок тяжкого ступеня.
Застосування у дітей:
Корекція дози у пацієнтів дитячого віку не потрібна.
Застосування у пацієнтів похилого віку:
Корекція дози у пацієнтів похилого віку (≥65 років) не потрібна.
Особливі вказівки:
У медичній документації пацієнта слід зазначати торгове найменування препарату Гемлібра® та номер серії.
Пацієнтам/особам, які доглядають за пацієнтами, слід рекомендувати записувати номер серії препарату Гемлібра® при його введенні поза медичним закладом.
Тромботична мікроангіопатія, пов'язана із застосуванням препарату Гемлібра® та активованого концентрату протромбінового комплексу (аКПК)
У клінічному дослідженні повідомлялося про явища ТМА у пацієнтів, які отримували профілактику препаратом Гемлібра®, при введенні середньої кумулятивної дози аКПК >100 Од/кг/24 год протягом ≥24 год. Лікування ТМА включало підтримуючу терапію з або без проведення плазмаферезу та гемоді. Ознаки, що підтверджують покращення, спостерігалися протягом одного тижня після припинення застосування АКПК. Таке швидке клінічне покращення не характерне для звичайного клінічного перебігу атипового гемолітико-уремічного синдрому та класичних ТМА, таких як тромботична тромбоцитопенічна пурпура.
За пацієнтами, які одночасно одержують профілактику препаратом Гемлібра® та аКПК, слід спостерігати на предмет розвитку ТМА. Лікар повинен негайно відмінити аКПК і перервати терапію препаратом Гемлібра® при виникненні клінічних симптомів та/або лабораторних показників, що відповідають ТМА, та провести лікування відповідно до клінічних показань. Після повного дозволу ТМА лікар і пацієнт/особа, що здійснює догляд за пацієнтом, повинні оцінити співвідношення користі та ризику відновлення профілактики препаратом Гемлібра на індивідуальній основі.
Якщо пацієнту, який отримує профілактику препаратом Гемлібра®, показаний препарат шунтуючої дії, див. нижче рекомендації з дозування препаратів шунтуючої дії (підрозділ "Рекомендації щодо застосування препаратів шунтуючої дії у пацієнтів, які отримують профілактику препаратом Гемлібра®").
Тромбоемболія, пов'язана із застосуванням препарату Гемлібра® та активованого концентрату протромбінового комплексу (аКПК)
У клінічному дослідженні повідомлялося про випадки розвитку тромботичних явищ у пацієнтів, які отримували профілактику препаратом Гемлібра®, при введенні середньої кумулятивної дози аКПК >100 Од/кг/24 год протягом ≥24 год. У жодному випадку не потрібно проведення антикоагулянтної терапії не характерно для нормальної тактики лікування тромботичних явищ. Ознаки покращення стану пацієнтів або розв'язання явищ спостерігалися після відміни аКПК.
За пацієнтами, які одночасно одержують профілактику препаратом Гемлібра® та аКПК, слід спостерігати на предмет розвитку тромбоемболії. Лікар повинен негайно відмінити аКПК та перервати терапію препаратом Гемлібра® при виникненні клінічних симптомів, отриманні даних візуалізуючих досліджень та/або лабораторних показників, що відповідають тромботичним явищам, та провести лікування відповідно до клінічних показань. Після повного вирішення тромботичного явища лікар і пацієнт/особа, що здійснює догляд за пацієнтом, повинні оцінити співвідношення користі та ризику відновлення профілактики препаратом Гемлібра на індивідуальній основі.
Якщо пацієнту, який отримує профілактику препаратом Гемлібра®, показаний препарат шунтуючої дії, див. наведені нижче рекомендації щодо дозування препаратів шунтуючої дії.
Рекомендації щодо застосування препаратів шунтуючої дії у пацієнтів, які отримують профілактику препаратом Гемлібра®
Лікування препаратами шунтуючої дії слід відмінити за день до початку терапії препаратом Гемлібра.
Лікарі повинні обговорювати точні дози та графік введення препаратів шунтуючої дії з усіма пацієнтами та/або особами, які доглядають за пацієнтами, якщо їх застосування потрібне під час профілактики препаратом Гемлібра®.
Препарат Гемлібра® підвищує здатність крові до зсідання. Отже, необхідна доза препарату шунтуючої дії може бути нижчою за таку, що використовується за відсутності профілактики препаратом Гемлібра®. Тривалість лікування препаратами шунтуючої дії та їх дозування залежатимуть від локалізації та об'єму кровотечі, а також від клінічного стану пацієнта.
Застосування АПК слід уникати, за винятком випадків, коли інші варіанти лікування/альтернативні засоби недоступні. Якщо пацієнту, який отримує профілактику препаратом Гемлібра®, показано застосування аКПК, початкова доза аКПК не повинна перевищувати 50 од./кг. Якщо кровотечу не вдається зупинити за допомогою початкової дози АКПК до 50 од./кг, слід ввести додаткові дози аКПК під керівництвом або наглядом медичного працівника, а загальна доза аКПК не повинна перевищувати 100 од./кг за перші 24 години лікування.
При розгляді питання про продовження терапії аКПК після введення максимальної дози 100 Од/кг протягом перших 24 годин лікарі повинні ретельно зіставити ризик розвитку ТМА та тромбоемболії та ризик кровотечі.
У клінічних дослідженнях не спостерігалося випадків ТМА або тромботичних явищ при використанні лише активованого рекомбінантного людського фактора VII (rFVIIa) у пацієнтів, які отримували профілактику препаратом Гемлібра.
Слід дотримуватися цих вказівок щодо дозування препарату шунтуючої дії як мінімум протягом 6 місяців після припинення профілактики препаратом Гемлібра®.
Імуногенність:
У невеликої кількості пацієнтів, які отримували препарат Гемлібра в клінічних дослідженнях, спостерігалися антитіла до еміцизумабу. Більшість пацієнтів з антитілами до еміцизумабу не відзначали зміни концентрації еміцизумабу в плазмі крові або збільшення числа кровотеч; однак нечасто (≥1/1000-<1/100) наявність нейтралізуючих антитіл до еміцизумабу з одночасним зниженням концентрації еміцизумабу могла бути пов'язана з втратою ефективності (див. розділи "Фармакологічна дія" та "Побічна дія"). У разі клінічних проявів втрати ефективності (наприклад, збільшення числа проривних кровотеч) слід негайно провести лікарську оцінку для визначення етіології та розглянути можливість зміни лікування.
Вплив на лабораторні показники згортання крові:
Препарат Гемлібра® спотворює результати клоттингових лабораторних аналізів, що ґрунтуються на внутрішньому шляху згортання, в т.ч. результати аналізу на активований час згортання (АВС), аналіз АЧТВ та всі аналізи, засновані на АЧТВ, зокрема на одноетапний аналіз активності FVIII (див. таблицю 3). В цілому, результати клоттингових лабораторних аналізів, заснованих на внутрішньому шляху згортання, не слід використовувати з метою моніторингу активності препарату Гемлібра®, визначення дози замісної терапії, що містять фактори згортання, або антикоагулянтних препаратів, або вимірювання титрів інгібіторів FVIII. Лабораторні аналізи, на результати яких впливає або не впливає застосування Гемлібру, наведено нижче.
Аналізи на згортання крові, на результати яких впливає або не впливає застосування препарату Гемлібра®
Результати, що спотворюють при застосуванні препарату Гемлібра:
- АЧТВ.
- Бетесда тести (клоттингові) для визначення титрів інгібіторів FVIII.
- Одноетапні, засновані на АЧТВ, аналізи одного з факторів зсідання крові (наприклад, активність FVIII).
- Заснований на АЧТП аналіз на стійкість до дії активованого протеїну С (APC-R).
- Активований час зсідання (АВС).
Результати, що не спотворюються при застосуванні препарату Гемлібра:
- Бетесда тести (хромогенний аналіз з використанням бичачих факторів згортання) для визначення титрів інгібіторів FVIII.
- Тромбіновий час.
- Одноетапні, засновані на вимірюванні протромбінового часу (ПВ), аналізи одного з факторів згортання крові.
- Хромогенні аналізи одного з факторів зсідання крові, за винятком FVIII*.
- Імунні аналізи (наприклад, ELISA, турбідиметричний метод).
- Генетичні аналізи факторів згортання (наприклад, аналіз фактора V Лейдена, протромбіну 20210).
* Важливі положення щодо хромогенних аналізів активності FVIII див. у розділі "Лікарська взаємодія".
Використання в педіатрії:
Ефективність та безпека препарату Гемлібра® у пацієнтів дитячого віку встановлено. Застосування препарату Гемлібра у пацієнтів дитячого віку з гемофілією А вивчалося в ході кількох клінічних досліджень, у яких брали участь підлітки (від 12 до <18 років), діти (від 2 до <12 років) та немовлята (від 1 місяця до <2 років) ). Результати оцінки безпеки та ефективності відповідали таким, що спостерігалися у дорослих. Мінімальні концентрації еміцизумабу в плазмі у рівноважному стані були порівняні у дорослих пацієнтів та пацієнтів дитячого віку при застосуванні еквівалентних доз, розрахованих за масою тіла.
Інструкції зі знищення невикористаного препарату або препарату зі строком придатності, що минув.
Попадання лікарського препарату у довкілля має бути зведене до мінімуму. Не слід утилізувати препарат за допомогою стічних вод або разом із побутовими відходами. Знищення невикористаного препарату або препарату з терміном придатності, що минув, повинно проводитися відповідно до локальних вимог.
Вплив на здатність до керування транспортними засобами та механізмами
Дані щодо впливу препарату Гемлібра® на здатність до керування транспортними засобами та роботу з машинами та механізмами відсутні.
Передозування:
Дані про передозування препарату Гемлібра обмежені. Випадкове передозування може призвести до гіперкоагуляції.
Пацієнтам, у яких відбулося випадкове передозування, слід негайно зв'язатися зі своїм лікарем. Необхідне ретельне спостереження таких пацієнтів.
Лікарська взаємодія:
Адекватних чи добре контрольованих досліджень лікарської взаємодії з препаратом Гемлібра не проводилось.
Досвід клінічного застосування свідчить про існування лікарської взаємодії між препаратом Гемлібра та аКПК.
Згідно з даними доклінічних досліджень при одночасному застосуванні рекомбінантного активованого фактора VII (rFVIIa) або FVIII з препаратом Гемлібра існує ймовірність гіперкоагуляції.
Еміцизумаб підвищує здатність крові до зсідання, таким чином, доза фактора зсідання, яка потрібна для досягнення гемостазу, може бути нижчою, ніж без профілактики препаратом Гемлібра®.
Вплив препарату Гемлібра на результати аналізів згортання крові.
Препарат Гемлібру заповнює кофакторну активність відсутнього активованого фактора VIII (FVIIIa) у теназному комплексі.
В рамках лабораторних аналізів згортання крові, що ґрунтуються на внутрішньому шляху згортання (наприклад, вимірювання АЧТВ), визначається загальний час згортання, який включає час, необхідний для активації FVIII (освіта FVIIIa) під дією тромбіну. При застосуванні препарату Гемлібра активація під дією тромбіну не потрібна, тому результатом таких аналізів буде надмірно вкорочений час згортання. Надмірно вкорочений час згортання (внутрішнім шляхом) згодом спотворюватиме результати всіх аналізів, заснованих на АЧТВ і призначених для визначення одного з факторів згортання крові, таких як одноетапний аналіз активності FVIII.
Однак результати аналізів одного з факторів згортання крові при використанні хромогенного або імунного методів не спотворюються на фоні застосування препарату Гемлібра, тому їх можна застосовувати для контролю параметрів згортання в ході терапії з урахуванням особливостей хромогенних аналізів активності FVIII, описаних нижче.
У набори для хромогенного аналізу активності FVIII можуть бути включені людські або бичачі коагуляційні білки. Набори, в яких використовуються людські фактори згортання, є чутливими до препарату Гемлібра®, проте при їх застосуванні клінічний гемостатичний потенціал препарату Гемлібра® може бути завищений. Навпаки, набори, в яких використовуються бичачі фактори згортання, не чутливі до препарату Гемлібра (не вимірюють його активність), і їх можна використовувати для контролю активності ендогенного або введеного FVIII, або для вимірювання рівня інгібіторів FVIII.
Препарат Гемлібра® зберігає активність у присутності інгібіторів FVIII і, таким чином, при використанні клоттингових тестів Бетесда для визначення функціонального інгібування FVIII будуть спостерігатися хибнонегативні результати. Замість них можна використовувати тест Бетесда з використанням хромогенного аналізу на основі бичачого FVIII, який не є чутливим до препарату Гемлібра®.
У зв'язку з тривалим Т1/2 препарату Гемлібра® вплив на результати аналізів зсідання крові може зберігатися протягом 6 місяців після введення останньої дози.
Умови зберігання препарату Гемлібра:
Препарат слід зберігати у картонній пачці для захисту від світла, у недоступному для дітей місці при температурі 2-8°С; не заморожувати; не струшувати.
Термін придатності препарату Гемлібра:
Термін придатності – 2 роки.
Не застосовувати після закінчення терміну придатності.